研究内容
プレB細胞性急性リンパ性白血病の発生機構
プレB細胞性急性リンパ性白血病(pre-B ALL)は小児のがんの中で発症率が最も高い病気です。現在では化学療法など治療法の進歩により寛解率は向上していますが、未だ完全治癒は容易ではありません。また、化学療法の副作用が患者に大変な苦痛を与えることも問題であり、白血病細胞特異的に作用する新たな治療法が待たれています。pre-B ALLにはE2A-PBX、TEL/AML1あるいはBCR-ABLといった染色体転座が見出されることがありますが、多くはまだ原因が不明です。この白血病の発がんの原因を明らかにすることは、その標的治療の開発にとって不可欠であります。
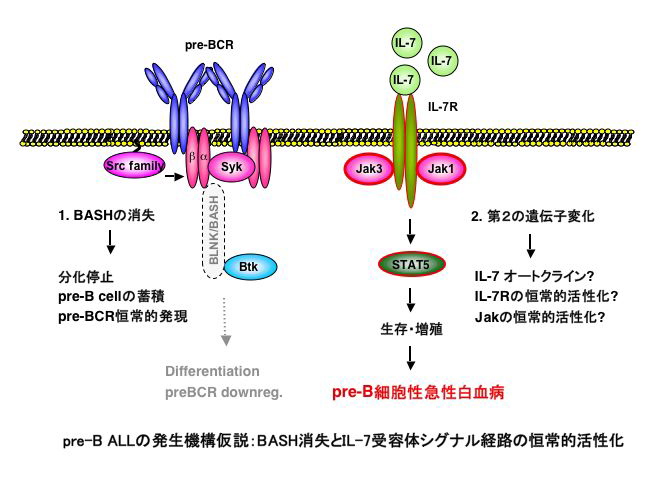
B細胞特異的アダプター蛋白 BLNK(SLP-65/BASH)は種々のシグナル因子と結合し、BCRシグナルによるカルシウム動員やMAPキナーゼ、NF-κB等の活性化、B細胞の生存・増殖に必要です。BLNK ノックアウト(KO)マウスではB細胞分化抑制が起こり、骨髄には普通では見られないプレB細胞受容体(preBCR)陽性の大型プレB細胞の蓄積がみられます。しかし、この細胞の細胞周期はG0/G1期で抑制されています。また、BCRの副受容体CD19とのダブルノックアウト(DKO)マウスではプレB細胞分化・増殖が完全に停止しました。したがって、部分的にCD19に代償されうるものの、BLNKはプレB細胞の増殖と分化を誘導するpreBCRシグナル伝達に必要であることが明らかになりました。驚いたことに、BLNK-KOマウスの約5%、BLNK/CD19-DKOマウスの約15%に、生後4週以内にpre-B ALLが発症しました。この白血病細胞は単クローン性で、転移性が高く、発症した個体は全部死亡しました(Hayashi et al., Immunity 2003)。ヒトにおいても、小児pre-B ALLの約半数例で白血病細胞内のBLNKの発現が消失していることが報告されています(Jumaa et al., Nature 2003)。したがって、BLNKはプレB細胞の癌化を抑制する癌抑制因子と考えられます。さらに、BLNKとBtkとの二重欠損マウスではプレB細胞分化の完全停止に加え、プレB細胞白血病の発症率が大幅に増加します(Kersseboom et al., JEM 2003)。CD19, SLP-76, Btkそれぞれ単独欠損マウスではプレB細胞分化抑制も白血病も起こらないので、これらはBLNK欠損下で部分的にBLNKの癌抑制機能を代償していると考えられます。
PreBCRを構成するμH鎖やλ5の欠損では白血病は発症しないので、BLNKを介する特異的シグナルがプレB細胞の癌化を抑制していると考えられます。また、発症率から考えてBLNK欠損に加えて多段階の遺伝子異常の蓄積が癌化に必要であると思われます。私たちはこのBLNK欠損マウスをモデルとして、また、発症したマウスから樹立したプレB白血病細胞を用いて、BLNKによる癌化抑制機構について研究してきました。その結果、BLNKがJak3に直接結合してその活性を抑制することで、IL-7受容体からSTAT5に至るシグナル伝達経路を抑制していることを明らかにしました。さらに、BLNK欠損マウス由来のプレB白血病細胞ではIL-7の異所性発現が見られました。以上より、何らかの体細胞突然によるIL-7の恒常的発現とBLNK欠損によるIL-7受容体シグナル伝達の恒常的活性化が、BLNK欠損マウスにおけるプレB白血病の成因と考えられました(Nakayama et al., Blood 2009)。この研究の成果は、プレB細胞の白血病化の分子機構を明らかにすると同時に、preBCRシグナル系を標的とした副作用の少ない治療法の開発に貢献すると期待されます。
Back